Tumor Heterogeneity Anaysis
Cancer is heterogeneous in the sense that the cancer cells in a tumor are not genetically identical, but form distinct clones, defined as subpopulations of cancer cells that host the same genomic aberrations. In aggressive and metastatic cancers, these genomic aberrations quickly evolve, resulting in extreme spatial and temporal heterogeneity (Shah et al. 2012, Nowell 1976). Therefore, multiple biopsies over different locations and at different time points need to be collected and sequenced in order to capture the complexity of tumor genomic landscapes and provide insight into how tumors evolve and escape treatment (Gerlinger et al. 2014, Frampton et al. 2013). Accordingly, computational tools are needed to accurately characterize the clonal structure of cancer and reveal how that structure evolves over time.
In recent years, a large number of computational tools and statistical models have been developed to analyze tumor heterogeneity from DNA sequencing data. However, most of these tools only model one type of genomic aberration, such as single-nucleotide variants (SNVs), copy number alterations (CNAs), or structural variants. Restricting the analysis to a single type of mutation not only reduces statistical power to accurately detect the clonal structure within the tumor, but also prevents us from understanding interactions among different types of mutations. Furthermore, many SNV-based methods assume that no copy number changes have occurred, which is extremely improbable. Therefore, their estimation of the prevalence of a given clone can be inaccurate, and the corresponding heterogeneity results may be misleading. Existing methods that capture SNVs and CNAs in the same model (i.e., phyloWGS from Deshwar et al. 2015, SPRUCE from El-Kebir et al. 2015, and Canopy from Jiang et al. 2016) require running a CNA-calling algorithm before heterogeneity analysis, but accurate CNA characterization also depends on heterogeneity analysis.
Most existing tools are designed to analyze a single tumor biopsy and are not suitable for jointly analyzing multiple biopsies. As DNA sequencing becomes more affordable, we can more easily collect multiple biopsies from a single patient during treatment. If we only perform heterogeneity analysis on the individual biopsies, then we are unable to detect clones that are shared across different biopsies from the same patient, and we fail to address important questions about how the tumor cells evolve, metastasize and escape treatment.
Finally, although most of models are free and publicly available, it is difficult to extend them by adding new assumptions and new types of biological data. Even under the best of circumstances, significant effort is required for users to fully understand the source code. In many situations, data structures and computational algorithms prohibit other investigators from modifying the model to accommodate their special needs.
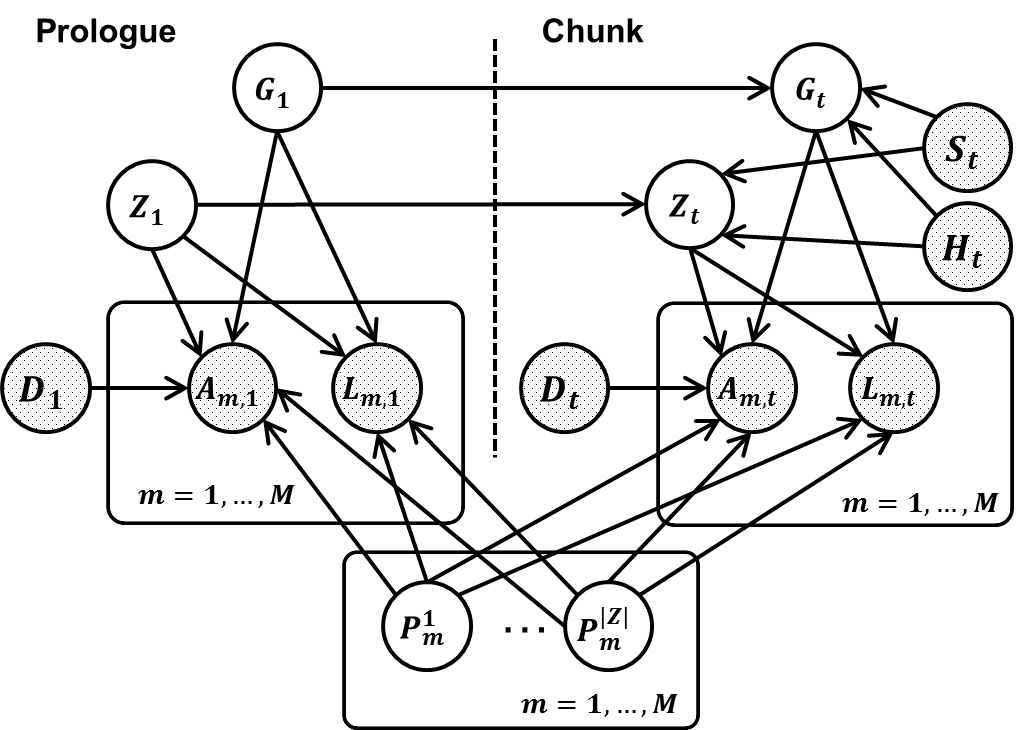
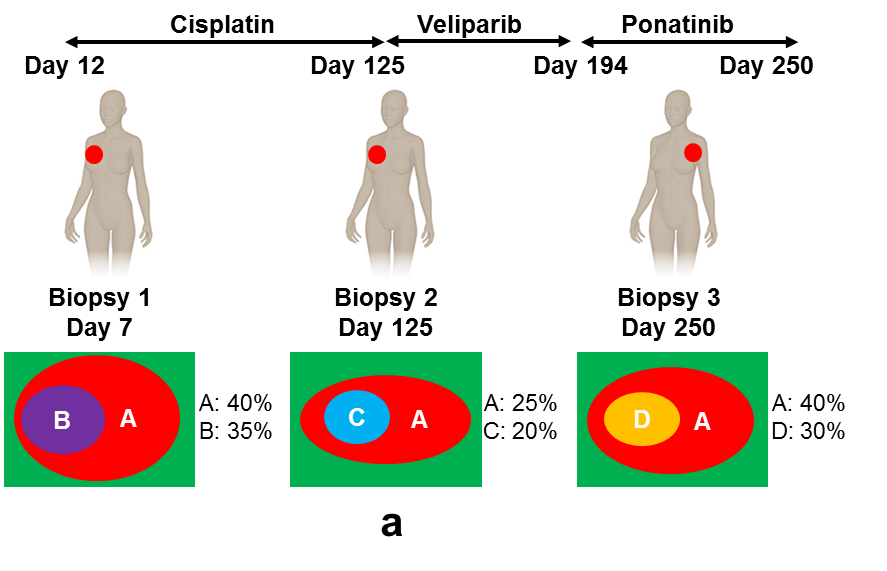
To address these challenges, we propose THEMIS (Tumor Heterogeneity Extensible Modeling via an Integrative System), which allows us to jointly characterize different types of genomic aberrations from multiple biopsies using a dynamic graphical model (in figure above). We implement our model via an extensible modeling platform, the Graphical Models Toolkit (GMTK) (Bilmes and Zweig 2002), which makes our approach open, reproducible and easy for others to extend. To extend the model, users only have to modify the model specification files; GMTK then automatically handles the required computation. Simulation experiments demonstrate that THEMIS significantly increases the accuracy of recovering tumor subclones and their genotypes, compared with its ancestor, TITAN (Ha et al. 2012). Single cell DNA sequencing confirms that individual nuclei can be segregated into one of the two tumor subclones identified by THEMIS. We applied THEMIS to three tumor biopsies from one cancer patient, thereby revealing the mutation accumulation history of the patient, tracking cancer progression, and identifying mutations related to developing resistance following various treatments.